LAMMPS
LAMMPS是由桑迪亚国家实验室开发的一套分子动力学模拟的开源程序包。LAMMPS使用MPI实现多机器并行计算,在新的版本中,支持基于CUDA和OpenCL的GPU计算。其以GNU通用公共许可证发布,因而开源自由。LAMMPS最初为一美国政府与私人机构合作项目,由美国能源部与另外三所私有企业实验室合作开发。
关于LAMMPS的更多信息请访问LAMMPS官网。
一、作业提交参数说明
用户可通过公共模板提交LAMMPS作业,与LAMMPS相关的作业参数如下:
| 参数 | 描述 |
| lammps file | 运行实例的源文件 |
| depend file | 实例运行的相关依赖参数文件 |
二、LAMMPS作业运行参考
1.修改运行文件in.melt
# 3d Lennard-Jones melt
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 10 0 10 0 10
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 3.0 87287
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify every 20 delay 0 check no
fix 1 all nve
dump id all atom 50 dump.melt
#dump 2 all image 25 image.*.jpg type type &
# axes yes 0.8 0.02 view 60 -30
#dump_modify 2 pad 3
#dump 3 all movie 25 movie.mpg type type &
# axes yes 0.8 0.02 view 60 -30
#dump_modify 3 pad 3
thermo 50
run 2500
2.执行命令
mpirun -np 4 lmp_mpi -in ~/lammps-install/examples/melt/in.melt
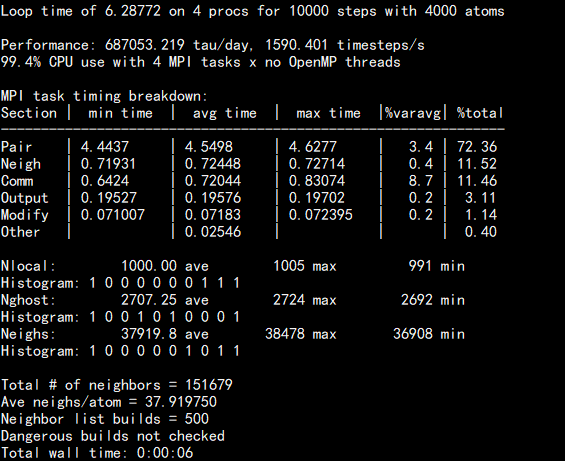
3.生成dump.melt文件
使用软件VMD(windows)可打开此文件

4.执行文件in.file
#Initial setup
dimension 3
units metal
boundary p p p
atom_style atomic
read_data data.file
mass 1 12.0108
mass 2 12.0108
mass 3 12.0108
# Set temperature
variable Teq equal 300
variable deltT equal 50
variable Th equal v_Teq+v_deltT
variable Tc equal v_Teq-v_deltT
#Initialize potential
pair_style hybrid/overlay tersoff lj/cut 10
pair_coeff * * tersoff BNC.tersoff C NULL NULL
pair_coeff 1 2 lj/cut 0.00456 3.431
pair_coeff 1 3 lj/cut 0.00456 3.431
pair_coeff 2 3 none
pair_coeff 2 2 none
pair_coeff 3 3 none
neighbor 2.0 nsq
neigh_modify every 1 delay 5 check yes
#Regions and group
region 1 block -0.1 4 EDGE EDGE EDGE EDGE units box
region 2 block 254 259 EDGE EDGE EDGE EDGE units box
region 3 block 4 12.5 EDGE EDGE EDGE EDGE units box
region 4 block 245.5 254 EDGE EDGE EDGE EDGE units box
region 5 block EDGE EDGE 31.7275 41.7275 EDGE EDGE units box
group GRA type 1
group probe1 type 2
group probe2 type 3
group probe union probe1 probe2
group f_1 region 1
group f_2 region 2
group GRA_heat region 3
group GRA_cool region 4
group Observe region 5
group f_x union f_1 f_2
group body subtract GRA f_1 f_2
timestep 0.0005
thermo 10000
dump 1 all xyz 100000 ALL.xyz
dump_modify 1 element C C C
dump 2 GRA xyz 100000 GRA.xyz
dump_modify 2 element C C C
dump 3 Observe xyz 100000 Obs.xyz
dump_modify 3 element C C C
#Initial velocity
velocity GRA create ${Teq} 92483151 mom yes rot yes dist gaussian
#fix the probe
velocity probe1 set 0.0 0.0 0.0 units box sum no
fix fp1 probe1 setforce 0.0 0.0 0.0
velocity probe2 set 0.0 0.0 0.0 units box sum no
fix fp2 probe2 setforce 0.0 0.0 0.0
compute Gtemp GRA temp
compute btemp body temp
#NPT1
fix npt1 GRA npt temp ${Teq} ${Teq} 0.05 iso 0.0 0.0 0.5 drag 0.5
fix_modify npt1 temp Gtemp
fix r1 all recenter INIT INIT INIT units box
thermo_modify temp Gtemp
run 1000000
unfix npt1
unfix r1
#fix the boundary
velocity f_1 set 0.0 0.0 0.0 units box
fix f1 f_1 setforce 0.0 0.0 0.0
velocity f_2 set 0.0 0.0 0.0 units box
fix f2 f_2 setforce 0.0 0.0 0.0
#NVT move the probe
fix nvt1 all nvt temp ${Teq} ${Teq} 0.05 drag 0.2
fix_modify nvt1 temp btemp
thermo_modify temp btemp
velocity probe1 set 0.0 0.0 -0.04 units box
run 250000
velocity probe set 0.0 0.0 -0.04 units box
run 1500000
velocity probe set 0.0 0.0 0.04 units box
run 250000
velocity probe set 0.0 0.0 0.0 units box
unfix nvt1
#NVE
fix nve GRA nve
thermo_style custom step temp
thermo_modify temp btemp
run 1000000
#NEMD
compute TGRAhot GRA_heat temp/com
compute TGRAcold GRA_cool temp/com
variable TGRAdif equal c_TGRAhot-c_TGRAcold
fix GRAhot GRA_heat langevin ${Th} ${Th} 0.01 25368 tally yes
fix GRAcold GRA_cool langevin ${Tc} ${Tc} 0.01 36368 tally yes
thermo_style custom step temp c_TGRAhot c_TGRAcold v_TGRAdif
thermo_modify temp btemp
run 8000000
unfix GRAhot
unfix GRAcold
# thermal conductivity calculation
# reset langevin thermostats to zero energy accumulation
reset_timestep 0
log result.log
compute ke_atom body ke/atom
fix GRAhot GRA_heat langevin ${Th} ${Th} 0.01 25368 tally yes
fix GRAcold GRA_cool langevin ${Tc} ${Tc} 0.01 36368 tally yes
variable time1 equal step*0.0005 # unit:ps
thermo_style custom step v_time1 temp c_TGRAhot c_TGRAcold v_TGRAdif f_GRAhot f_GRAcold
thermo_modify temp btemp
compute cc1 body chunk/atom bin/1d x lower 2 units box
fix average body ave/chunk 1 200 200 cc1 c_ke_atom file ke_atom.txt
fix ave body ave/time 1 200 200 f_GRAhot f_GRAcold file Energy.txt
run 2000000
print "Program successfully finish!"
5.依赖文件
data.file BNC.tersoff
6.input文件
#job_name=lmp
#run_time=24:00:00
partition=dell_intel
node_num=3
task_per_node=32
lammmps_file=/home/wushiming/lammps/in.file
depend_file=(/home/wushiming/lammps/data.file /home/wushiming/lammps/BNC.tersoff)
#work_dir=/home/wushiming/lammps
7.执行脚本
#!/bin/sh
source /home/wushiming/lammps/lammps_input
##check input var
time=`date +%m%d_%H%M%S`
if [ "x$job_name" == "x" ];then
sbatch_job_name="YHPC_$time "
else
sbatch_job_name=$job_name
fi
if [ "x$partition" == "x" ];then
sbatch_partition=""
else
sbatch_partition=$partition
fi
if [ "x$work_dir" == "x" ];then
mkdir -p /home/yhpc/YHPC_$time
sbatch_work_dir=/home/yhpc/YHPC_$time
else
sbatch_work_dir=$work_dir
fi
if [ "x$run_time" == "x" ];then
sbatch_run_time=03:00:00
else
sbatch_run_time=$run_time
fi
if [ "x$lammmps_file" == "x" ];then
echo "The analysis_file cannot be empty."
exit 1
else
sbatch_lammps_file=$lammmps_file
fi
if [ "x$depend_file" == "x" ];then
continue
else
for i in ${depend_file[*]};do cp $i $sbatch_work_dir;done
fi
sbatch_node_num=$node_num
sbatch_task_per_node=$task_per_node
sbatch_err_log=$sbatch_work_dir/%j.err
sbatch_out_log=$sbatch_work_dir/%j.out
### Write basic job infomations
#echo -e "The start time is: `date +"%Y-%m-%d %H:%M:%S"` \n"
#echo -e "My job ID is: $SLURM_JOB_ID \n"
#echo -e "The total cores is: $total_cores \n"
#echo -e "The hosts is: \n"
#srun -np $node_num -nnp 1 hostname
cat > $sbatch_work_dir/lammps.slurm <<EOF
#!/bin/bash
#SBATCH --ntasks-per-node=$sbatch_task_per_node
#SBATCH --job-name $sbatch_job_name
#SBATCH --nodes=$sbatch_node_num
#SBATCH --mail-type=ALL
#SBATCH --partition $sbatch_partition
#SBATCH --chdir=$sbatch_work_dir
#SBATCH -e $sbatch_err_log
#SBATCH -o $sbatch_out_log
ulimit -s unlimited
ulimit -l unlimited
# 导入运行环境
module purge
source /opt/ohpc/pub/apps/intel/setvars.sh
module load intel/mpi-2021.1.1
module load gnu8/8.3.0
module load lammps/intel-lammps
export I_MPI_OFI_PROVIDER=Verbs
export FI_VERBS_IFACE=team1.282
echo -e "The start time is: `date +"%Y-%m-%d %H:%M:%S"`"
echo -e "My job ID is: SLURM_JOB_ID"
echo -e "The total cores is: SLURM_NPROCS"
echo -e "The SLURM_JOB_ID Job info:"
scontrol show job SLURM_JOB_ID
mpirun -genv I_MPI_FABRICS ofi lmp -in $lammmps_file
echo -e "The end time is: `date +"%Y-%m-%d %H:%M:%S"` \n"
EOF
sed -i 's/SLURM*/\$SLURM/g' $sbatch_work_dir/lammps.slurm
/usr/bin/sbatch $sbatch_work_dir/lammps.slurm